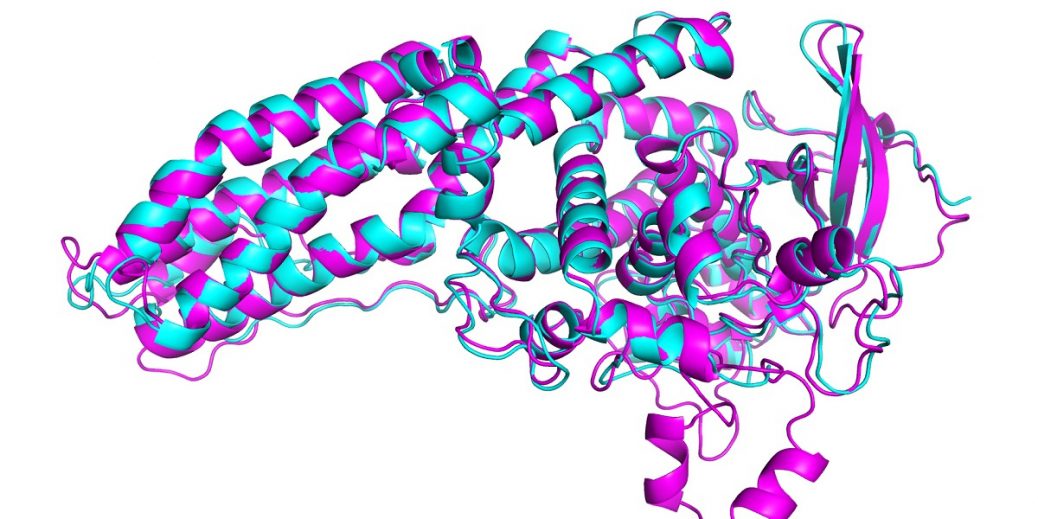
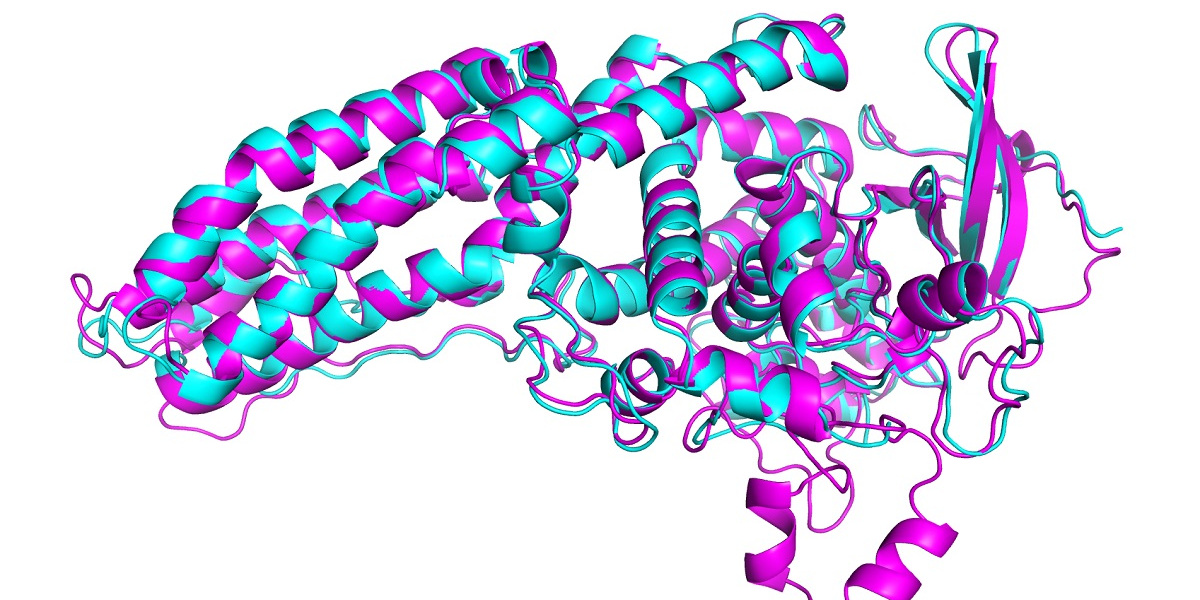
In this year’s CASP, AlphaFold predicted the structure of dozens of proteins with a margin of error of just 1.6 angstroms—that’s 0.16 nanometers. This far outstrips all other computational methods and for the first time matches the accuracy of experimental techniques to map out the structure of proteins in the lab, such as cryo-electron microscopy, nuclear magnetic resonance and x-ray crystallography. These techniques are expensive and slow: it can take hundreds of thousands of dollars and years of trial and error for each protein. AlphaFold can find a protein’s shape in a few days.
But identifying a protein’s structure is very hard. For most proteins, researchers have the sequence of amino acids in the ribbon but not the contorted shape they fold into. And there are typically an astronomical number of possible shapes for each sequence. Researchers have been wrestling with the problem at least since the 1970s, when Christian Anfinsen won the Nobel prize for showing that sequences determined structure.
The breakthrough could help researchers design new drugs and understand diseases. In the longer term, predicting protein structure will also help design synthetic proteins, such as enzymes that digest waste or produce biofuels. Researchers are also exploring ways to introduce synthetic proteins that will increase crop yields and make plants more nutritious.
Shocking advance
“It’s a very substantial advance,” says Mohammed AlQuraishi, a systems biologist at Columbia University who has developed his own system for predicting protein structure. “It’s something I simply didn’t expect to happen nearly this rapidly. It’s shocking, in a way.”
“This really is a big deal,” says David Baker, head of the Institute for Protein Design at the University of Washington and leader of the team behind Rosetta, a family of protein analysis tools. “It’s an amazing achievement, like what they did with Go.”
The launch of CASP in 1994 gave the field of predicting protein structures a boost. Every two years, the organizers release 100 or so amino acid sequences for proteins whose shapes have been identified in the lab but not yet made public. Dozens of teams from around the world then compete to find the correct way to fold them up using software. Many of the tools developed for CASP are already used by medical researchers. But progress was slow, with two decades of incremental advances failing to produce a shortcut to painstaking lab work.
When DeepMind entered the competition in 2018 with its first version of AlphaFold, it gave CASP the jolt it was looking for. It still could not match the accuracy of a lab but it left other computational techniques in the dust. Researchers took note: soon many were adapting their own systems to work more like AlphaFold.
This year more than half of the entries use some form of deep learning, says Moult. The accuracy overall was higher as a result. Baker’s new system, called trRosetta, uses some of DeepMind’s ideas from 2018. But it still came a “very distant second,” he says.
In CASP, results are scored using what’s known as a global distance test (GDT), which measures on a scale from 0 to 100 how close a predicted structure is to the actual shape of a protein identified in lab experiments. The latest version of AlphaFold scored well for all proteins in the challenge. But it got a GDT score above 90 for around two thirds of them. Its GDT for the hardest proteins was 25 points higher than the next best team, says John Jumper, who heads up the AlphaFold team at DeepMind. In 2018 the lead was around six points.



{kind=link}
Recent Comments